


Researchers have uncovered a new principle that could accelerate the development of cheaper and more efficient fuel cells by revealing how dual-atom catalysts behave during a key energy conversion reaction. The study, led by researchers at Tohoku University, shows that these catalysts follow a previously unknown “dual-Sabatier optima” pattern, overturning long-standing assumptions in catalyst science.
Details of the findings were published on April 27, 2026.
Fuel cells are widely seen as an important technology for building a low-carbon society because they can generate electricity cleanly from hydrogen. However, many fuel cells still rely on expensive precious metals such as platinum to drive the oxygen reduction reaction (ORR), a critical step that strongly influences performance and cost.
In recent years, scientists have explored atomically dispersed catalysts as a promising alternative. While single-atom catalysts contain isolated metal atoms, dual-atom catalysts (DACs) use pairs of atoms working together. Experiments have repeatedly shown that DACs can outperform single-atom systems, but researchers did not fully understand why.
Traditionally, scientists used a “single-peak volcano” model to explain catalytic activity. According to this idea, catalysts perform best only within a narrow range of chemical properties. However, when the research team analyzed large-scale experimental data from the Digital Catalysis Platform (DigCat)![]() , they found that the behavior of DACs did not match this conventional picture.
, they found that the behavior of DACs did not match this conventional picture.
To investigate further, the team systematically studied more than 200 dual-atom catalysts using advanced theoretical simulations, microkinetic modeling, and machine learning techniques. Their results revealed that DACs are mainly governed by a different reaction pathway known as the dissociative mechanism, rather than the associative mechanism commonly seen in single-atom catalysts.
This difference fundamentally changes how catalytic activity is distributed. Instead of one optimal peak, the researchers discovered two distinct optimal regions — a phenomenon they call “dual-Sabatier optima.” The team showed that these two activity peaks emerge because the rate-limiting step changes during the reaction process, switching among oxygen dissociation, oxygen protonation, and hydroxyl protonation steps.
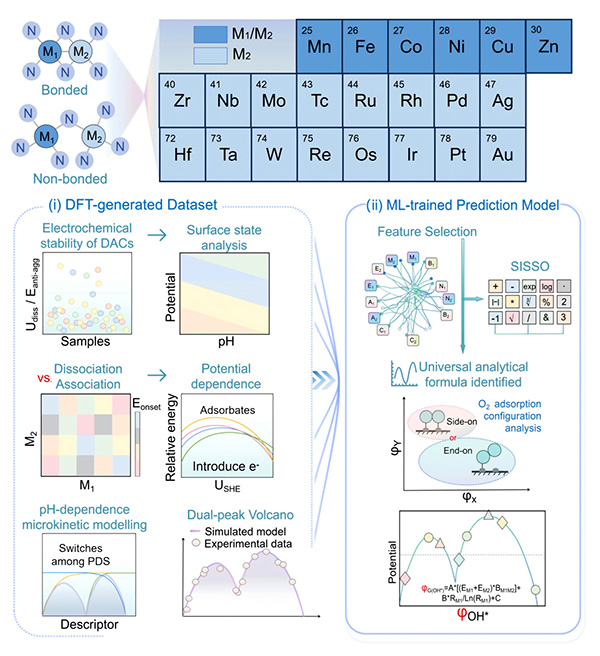
General workflow of this work, including two major parts: (i) Establish a DFT-generated dataset, and (ii) A ML prediction model based on structural descriptors. ©Jin Liu et al.
The researchers also demonstrated that the new principle applies broadly across many different catalyst systems, including those containing transition metals, metal-like elements, and even non-metal atoms. By combining interpretable machine learning with theoretical modeling, the team created a predictive framework that can rapidly identify promising catalyst structures with high accuracy.
“This discovery changes the way we think about catalyst design,” said Hao Li, Distinguished Professor at Tohoku University’s Advanced Institute for Materials Research (WPI-AIMR). “For a long time, researchers assumed that dual-atom catalysts followed the same activity rules as single-atom catalysts. Our work shows that entirely different mechanisms can emerge when two atoms cooperate together, opening new opportunities for designing highly efficient materials for clean energy technologies.”

Microkinetic ORR volcano models of M1M2-N-C DACs and rate-determining analyses. ORR activity as function of ΔG(OH*) contributed from dissociation and association mechanism over DACs at 0.9 V vs. RHE with (a) pH=1 and (b) pH=13 from theoretical calculation. (c) Experimental ORR activity as function of ΔG(OH*) over DACs at 0.9 V vs. RHE with pH=1 and pH=13. (d) The degree of rate control for each elementary step of dissociation mechanism as a function of ΔG(OH*) on DACs at 0.9 V vs. RHE. And Rate-determining analyses of the ORR process through dissociation mechanism. Dashed lines indicate the ORR activity solved by PDS analysis with the rate limited by the O2 protonation dissociation (blue), one of 2OH* protonation (red) and OH* protonation (black). ©Jin Liu et al.
Beyond fuel cells, the researchers believe the findings could influence the development of catalysts used in many other energy conversion and chemical production processes. The study also highlights how artificial intelligence can help scientists uncover hidden scientific principles from existing experimental data, dramatically shortening the time needed to discover new materials.
Moving forward, the team plans to expand their approach to more complex multimetallic catalyst systems and additional energy-related reactions beyond ORR. By integrating AI agents, machine learning, and electrochemical simulations into the DigCat platform, they hope to build a fully autonomous digital framework capable of rapidly designing next-generation catalysts for sustainable energy applications.

ML model prediction for potential M1A2-DACs. ©Jin Liu et al.
| タイトル: | Dual-Sabatier Optima: How Reaction Mechanism Determines Activity Volcano Map of Dual-Atom Catalysts for Oxygen Reduction Reaction |
|---|---|
| 著者: | Jin Liu, Hao Li, Haoxiang Xu and Daojian Cheng |
| 掲載誌: | Angewandte Chemie International Edition |
| DOI: | 10.1002/anie.8386838 |
東北大学材料科学高等研究所(WPI-AIMR)
教授 Hao Li(研究者プロフィール)
| E-mail: | li.hao.b8@tohoku.ac.jp |
|---|---|
| Webstie: | Hao Li Laboratory |
東北大学材料科学高等研究所(WPI-AIMR) 広報戦略室
| Tel: | 022-217-6146 |
|---|---|
| E-mail: | aimr-outreach@grp.tohoku.ac.jp |
