


06/25/2012
© 2012 APS
An important characteristic critical to a variety of technologies is the way in which oxide materials trap charges. For example, hafnium dioxide — used to insulate transistor gate electrodes — can begin to fail if charges become trapped in its bulk. Similarly, the effectiveness of zirconium dioxide as a common catalyst depends on the quantity of trapped charges. Accurately predicting the nature of charge trapping in oxides like these, however, has remained an unmet challenge. Now, Keith McKenna and Alexander Shluger from the AIMR at Tohoku University, along with colleagues from the UK and the USA, have developed a new calculational approach to predict unexpected aspects of charge localization — and thus trapping — in oxides1.
Calculations of charge trapping generally rely on density functional theory (DFT). The widely used implementations of DFT, however, characterize electrons as interacting with themselves electrostatically — a physically incorrect description. This ‘self-interaction error’ leads to inaccurate predictions of charge trapping in materials. McKenna, Shluger and colleagues have now carried out calculations using a version of density functional theory that includes cancellation of nonlinearity — that is, a correction to the self-interaction problem. This new insight provides an unprecedented view into how charges are localized in hafnium oxide and zirconium oxide.
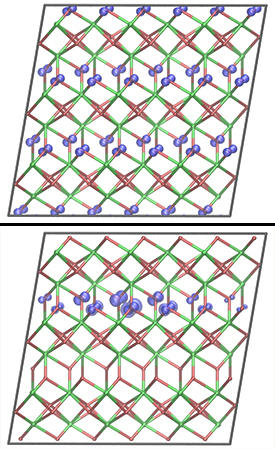
Although usually considered to be three-dimensional bulk crystals, the researchers’ calculations predicted that charges in these materials behave as if they were constrained to two dimensions. This is because the crystal structure of each material consists of alternating layers, in which oxygen atoms coordinate — or bond — to either three or four metal atoms. When a positive charge is induced in the materials, it induces distortions in the surrounding crystal lattice, and in turn quickly stabilizes the lattice structure (see image). The positive charge together with the lattice distortions are referred to as a polaron.
The researchers found that polarons prefer to stay in oxide layers with triply coordinated oxygen atoms. Such two-dimensional behavior has been much studied due to the particular physics it involves, but it has typically been observed only in complex oxides composed of three or more types of atoms, or at interfaces between materials. Its observation in a relatively simple binary oxide, made of only two types of atoms, is unexpected and will lead to a new investigative approach of the particular physics involved. “In addition to the importance of the theoretical methods themselves, our predictions may stimulate new studies of the correlated dynamics and interaction of positive charges in binary oxides,” says McKenna.
McKenna, K. P., Wolf, M. J., Shluger, A. L., Lany, S. & Zunger, A. Two-dimensional polaronic behavior in the binary oxides m-HfO2 and m-ZrO2. Physical Review Letters 108, 116403-1–116403-5 (2012). | article
This research highlight has been approved by the authors of the original article and all information and data contained within has been provided by said authors.

